BenchmarkST: Cross-Platform, Multi-Species Spatial Transcriptomics Gene Imputation Benchmarking
- Topics: bioinformatics, spatial transcriptomics, gene imputation, benchmarking, cross-platform/species analysis
- Skills:
- Programming Languages:
- Proficient in Python and/or R, commonly used in bioinformatics.
- Data Analysis:
- Experience with statistical data analysis and machine learning models.
- Bioinformatics Knowledge (not required but preferred):
- Proficiency in bioinformatics and computational biology.
- Familiarity with spatial transcriptomics datasets and platforms.
- Programming Languages:
- Difficulty: Advanced
- Size: Large (350 hours). Given the scope of integrating multi-platform, multi-species datasets and the complexity of benchmarking gene imputation methods, this project is substantial. It requires extensive data preparation, analysis, and validation phases, making it suitable for a larger time investment.
- Mentors: Ziheng Duan (contact person)
Project Idea Description
The orchestration of cellular life is profoundly influenced by the precise control of gene activation and silencing across different spatial and temporal contexts. Understanding these complex spatiotemporal gene expression patterns is vital for advancing our knowledge of biological processes, from development and disease progression to adaptation. While single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to profile gene expression across thousands of cells simultaneously, its requirement for cell dissociation strips away the critical spatial context, limiting our comprehension of cellular interactions within their native environments. Recent strides in spatial transcriptomics have started to bridge this gap by enabling spatially resolved gene expression measurements at single-cell or even sub-cellular resolutions. These advancements offer unparalleled opportunities to delineate the intricate tapestry of gene expression within tissues, shedding light on the dynamic interactions between cells and their surroundings.
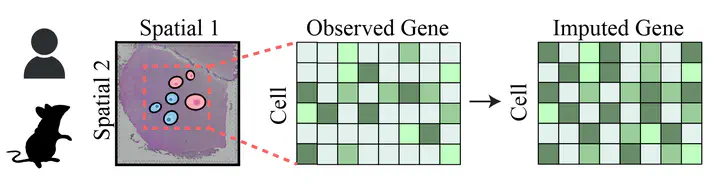
Despite these technological advances, a significant challenge remains: the datasets generated by spatial transcriptomic technologies are often incomplete, marred by missing gene expression values due to various technical and biological constraints. This limitation severely impedes our ability to fully interpret these rich datasets and extract meaningful insights from them. Gene imputation emerges as a pivotal solution to this problem, aiming to fill in these missing data points, thereby enhancing the resolution, quality, and interpretability of spatial transcriptomic datasets.
Recognizing the critical importance of this task, there is a pressing need for a unified benchmarking platform that can facilitate the evaluation and comparison of gene imputation methods across a diverse array of samples, spanning multiple sampling platforms, species, and organs. Currently, the bioinformatics and spatial transcriptomics fields lack such a standardized framework, hindering progress and innovation. To address this gap, our project aims to establish a comprehensive gene imputation dataset that encompasses a wide range of conditions and parameters. We intend to reproduce known methods and assess their efficacy, providing a solid and reproducible foundation for future advancements in this domain.
Project Deliverable
- A comprehensive, preprocessed benchmark dataset that spans multiple sampling platforms, species, and organs, aimed at standardizing gene imputation tasks in spatial transcriptomics.
- An objective comparison of state-of-the-art gene imputation methodologies, enhancing the understanding of their performance and applicability across diverse biological contexts.
- A user-friendly Python package offering a suite of gene imputation tools, designed to fulfill the research needs of the spatial transcriptomics community by improving data completeness and reproducibility.
Ziheng Duan
Ph.D. Student, University of California, Irvine
My research interests include computational biology and machine learning.